Von Genen zu Organismen: Bioinformatische Systemmodelle
und Software
Genregulatorische Netzwerke spielen eine zentrale Rolle bei der Entwicklung und Gesundheit jedes Organismus. Die Entschlüsselung und Modellierung dieser Netzwerke ist von vorrangiger Bedeutung für ein besseres Verständnis von Entwicklungsprozessen und genetisch bedingten Krankheiten. Das Ziel meiner Doktorarbeit war es
entwicklung eines umfassenden Rahmens für das Reverse Engineering von genregulatorischen Netzwerken
.
Die Bausteine dieses Frameworks wurden so konzipiert, dass sie erweiterbar sind, um eines Tages das Reverse Engineering ganzer Organismen zu ermöglichen:
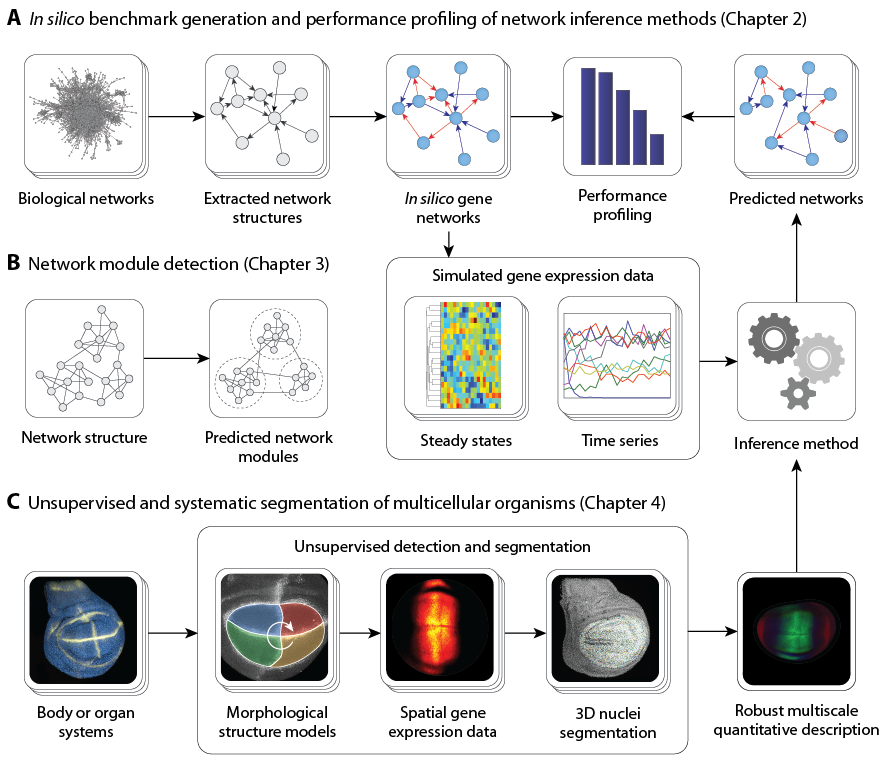
Biologische Interaktionsnetzwerke sind oft in Gruppen (auch Cluster, Module oder Gemeinschaften genannt) verwandter Gene und Proteine organisiert, die bestimmte biologische Funktionen erfüllen. Wir haben eine
konsensbasierte Community-Erkennungsmethode zur zuverlässigen Modulerkennung in Netzwerken
. Die Software kann auch verwendet werden, um Module in metabolischen, neuronalen, sozialen (z. B. LinkedIn, Facebook usw.) Bereichen zu identifizieren.) und technologische Netzwerke.
Zusätzlich zu den oben genannten
forschungsbeitrag
wurden die in dieser Arbeit entwickelten Methoden implementiert und veröffentlicht als
quelloffene, erweiterbare und benutzerfreundliche Softwareanwendungen
. Als Beispiel,
GennEtzWerker (GNW)
hat sich zu einem Standardwerkzeug für die Benchmarkgenerierung und Leistungsprofilierung von Gennetzwerk-Inferenzmethoden entwickelt. Bis heute hat die Community GNW verwendet, um die Genauigkeit von zu bewerten
5.000 Gennetzwerkvorhersagen
. Darüber hinaus wurde GNW verwendet, um drei Ausgaben der zu organisieren
TRAUM Herausforderung
, eine jährliche gemeinschaftsweite Netzwerk-Inferenz-Herausforderung.
Auf dem Weg zur unbeaufsichtigten und systematischen Segmentierung biologischer Organismen
Erhebliche Anstrengungen wurden unternommen, um genregulatorische Netzwerke aus Proteinkonzentrationsniveaus, die in einer einzelnen Zelle des Organismus gemessen werden können (DNA-Mikroarray-Daten), rückzuentwickeln. Die Rekonstruktion von Gennetzwerken ist jedoch in der Regel ein unterbestimmtes Problem, dh das echte Gennetzwerk kann nicht identifiziert werden
aufgrund fehlender verwertbarer Informationen
.
Bildgebungsanwendungen für die Hochdurchsatzmikroskopie stellen ein wichtiges Forschungsgebiet dar, das eines Tages Werkzeuge für die automatische Quantifizierung lebender Organismen auf Multiskalensystemebene (molekular-, Zell- und Gewebeebene) bereitstellen wird. In diesem Projekt haben wir eine entwickelt
verfahren zur unüberwachten und systematischen Segmentierung der
Drosophila
Flügel
. Diese Methode wird als Open-Source-, benutzerfreundliche und erweiterbare Software namens veröffentlicht
WingJ
.
Wir haben eine Methode zur Generierung von
quantitative Beschreibungen
von mehrzelligen Systemen (wie Körper- oder Organsystemen) aus 3D-Mikroskopbildern. Die quantitative Beschreibung berücksichtigt
informationen zu Morphologie, Genexpression und Zellkernen
.
Eine robuste und zuverlässige quantitative Beschreibung erfordert die Quantifizierung zahlreicher Einzelsysteme. In diesem Projekt haben wir eine entwickelt
unbeaufsichtigter Bildsegmentierungsalgorithmus zur schnellen und systematischen Quantifizierung der morphologischen Struktur des
Drosophila
Flügel
. Die Segmentierung der
Drosophila
embryonen werden ebenfalls unterstützt.
Wir haben unsere Methode angewendet, um Hunderte von zu segmentieren
Drosophila
flügel, die zu verschiedenen Zeitpunkten ihrer Entwicklung und für unterschiedliche genetische Hintergründe abgebildet wurden. Die generierte quantitative Beschreibung bietet ein leistungsfähiges Werkzeug für
besseres Verständnis der Biologie des Flügels und quantitative Bewertung der Auswirkungen von Medikamenten oder Mutationen
.
Erkennung von Gemeinschaftsstrukturen in komplexen Netzwerken
Biologische Interaktionsnetzwerke sind oft in Gruppen (auch Cluster, Module oder Gemeinschaften genannt) verwandter Gene und Proteine organisiert, die bestimmte biologische Funktionen erfüllen. Community-Erkennung hat zahlreiche Anwendungen für Systeme, die als Graphen beschrieben werden können, zum Beispiel
metabolische, neuronale, soziale (z.B. Facebook) und technologische Netzwerke
.
Jmod ist eine Open-Source-Java-Bibliothek für
erkennung von Gemeinschaftsstrukturen in Netzwerken
das kann leicht in Anwendungen von Drittanbietern integriert werden. Jmod implementiert mehrere Algorithmen, darunter einen neuartigen
konsensus-Community-Erkennungsmethode
die wir zur Identifizierung von Funktionsmodulen in transkriptionellen Netzwerken entwickelt haben. Jmod ist auch als eigenständige Anwendung mit grafischen Benutzeroberflächen und Befehlszeilenoberflächen verfügbar.
Das zweite Ziel dieses Projekts ist es, eine intuitive und vollständige Umgebung für die Entwicklung neuartiger Community-Erkennungsmethoden bereitzustellen. Jmod implementiert mehrere
benchmarks und Metriken
um die Leistung dieser Methoden zu bewerten. Eine Vielzahl zusätzlicher Tools ermöglicht es Forschern
fokus auf die Entwicklung neuartiger Methoden
und verbringen Sie weniger Zeit mit allgemeinen Aspekten der Community-Erkennung (z. B. Lesen von Netzwerkstrukturen, Implementieren von Standardmetriken usw.).).
In silico
Benchmarkgenerierung und Leistungsprofilierung von Netzwerkinferenzmethoden
Es wurden zahlreiche Methoden entwickelt, um genregulatorische Netzwerke aus Expressionsdaten zurückzuentwickeln. Die Entschlüsselung und Modellierung dieser Netzwerke ist von vorrangiger Bedeutung für ein besseres Verständnis biologischer Organismen. Sowohl ihre absolute als auch ihre vergleichende Leistung sind jedoch noch wenig verstanden.
Ziel dieses Projekts ist es, Benchmarks und Werkzeuge für die rigorose Erprobung von Methoden zur Gennetzwerkinferenz bereitzustellen.
Unser Framework ist verfügbar als
quelloffene und benutzerfreundliche Softwareanwendung namens GeneNetWeaver (GNW)
. GNW ist das erste Tool, das Methoden für beide bereitstellt
in silico
benchmark-Generierung und Leistungsprofilierung von Netzwerkrekonstruktionsalgorithmen.
Detaillierte Modelle von genregulatorischen Netzwerken
werden mit nur wenigen Klicks generiert. Ihre Struktur wird aus bekannten Transkriptionsnetzwerken extrahiert (
E. coli
,
S. cerevisiae
, usw.), bevor er mit detaillierten dynamischen Modellen der Genregulation ausgestattet wurde, die sowohl Transkription als auch Translation, unabhängige und synergistische Wechselwirkungen sowie molekulares Rauschen und Messrauschen berücksichtigen.
Wir haben GNW genutzt, um drei Ausgaben der zu organisieren
TRAUM Herausforderung
, eine jährliche gemeinschaftsweite Netzwerk-Inferenz-Herausforderung. In diesem Zusammenhang wurde GNW verwendet, um zu identifizieren
systematische Fehler
von Netzwerkinferenzalgorithmen und liefert so nützliche Einblicke in
wie Sie ihre Leistung verbessern können
.
Beobachtung und Interaktion in experimentellen Umgebungen
Die Unterscheidung von Subpopulationen in Gruppenverhaltensexperimenten kann den Einfluss von Unterschieden in der genetischen, pharmakologischen und Lebensgeschichte auf soziale Interaktionen und Entscheidungsfindung aufdecken. Wir haben entwickelt
Fluoreszenz-Verhaltens-Bildgebung (FBI)
, ein Toolkit, das transgene Fluoreszenz zur Unterscheidung von Subpopulationen verwendet, Bildgebungshardware, die gleichzeitig Verhalten und Fluoreszenzexpression aufzeichnet, und Open-Source-Software für die automatisierte, hochgenaue Bestimmung der genetischen Identität.
Das effektive Reverse Engineering von genregulatorischen Netzwerken ist eine der großen Herausforderungen der Systembiologie und wird voraussichtlich
erhebliche Auswirkungen auf die Pharma- und Biotech-Industrie in den nächsten Jahrzehnten
. Ein Gennetzwerk wird durch regulatorische Gene gebildet, die für Proteine kodieren, die die Expression anderer regulatorischer und / oder nicht-regulatorischer Gene verstärken oder hemmen, wodurch ein komplexes Netz von Wechselwirkungen gebildet wird. Ziel des Reverse Engineering ist es, ein solches Netzwerk aus experimentellen Daten automatisch zu identifizieren. In diesem Projekt,
Ich habe eine entwickelt
reverse-Engineering-Algorithmus, der in der Lage ist, Vorhersagemodelle von Gennetzwerken zu generieren
. Diese Modelle sind biologisch plausible dynamische Modelle, die verwendet werden können, um die Reaktion der Gennetzwerke auf neue Störungen wie die Anwendung eines Arzneimittels vorherzusagen;
Ich habe die Initiative ergriffen und eine Methode entwickelt für
erzeugende
in silico
benchmark von Netzwerken und Bewertung der Leistung von Reverse-Engineering-Algorithmen
. Diese Arbeit inspirierte später die Entwicklung der
Genetzwerker
Projekt;
Ich habe das erhalten
Preis für das beste Poster
auf der Ausstellung der Meisterprojekte des Mikroingenieurs 2008 (mehr als 90 Teilnehmer).